Output Generated¶
This summarizes the expected output depending on how the pipeline logic boolean values are set in the config file.
Full Pipeline with Chunking¶
Setting this logic:
GenerateGRM:true
GenerateNull:true
GenerateAssociations:true
GenerateResults:true
SkipChunking:false
SaveChunks:true
Returns this output:

What can be reused?
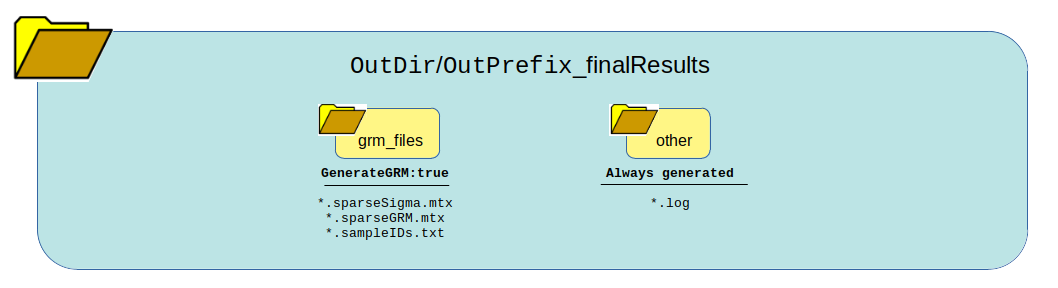
Genetic Relatedness Matrix (GRM) only¶
Setting this logic:
GenerateGRM:true
GenerateNull:false
GenerateAssociations:false
GenerateResults:false
SkipChunking:false
Returns this output:
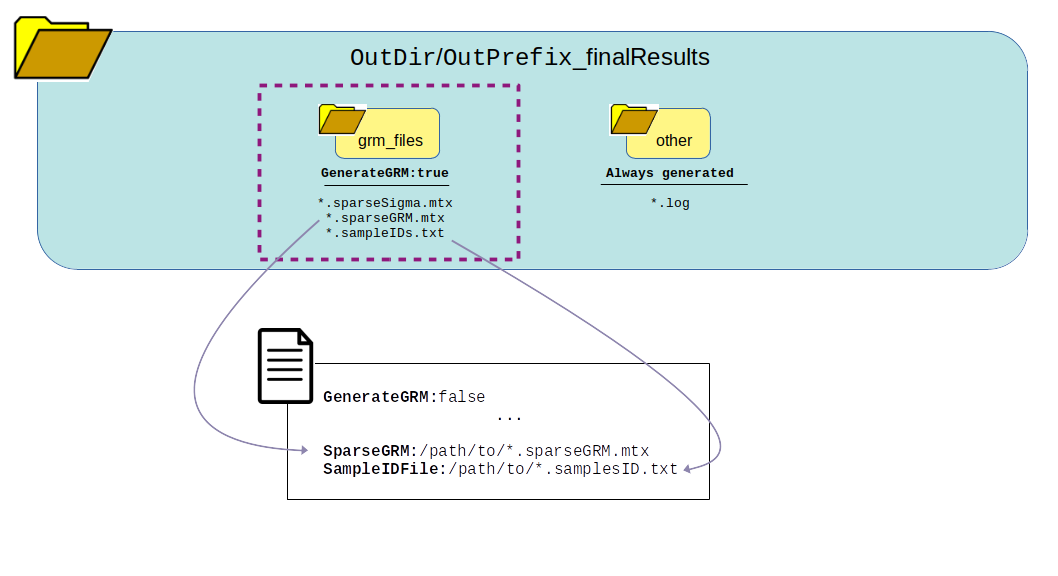
What can be reused?
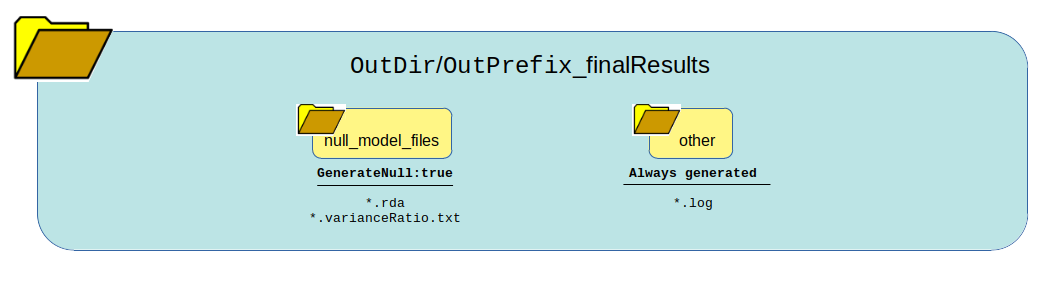
Null Model only¶
Setting this logic:
GenerateGRM:false
GenerateNull:true
GenerateAssociations:false
GenerateResults:false
SkipChunking:false
Returns this output:
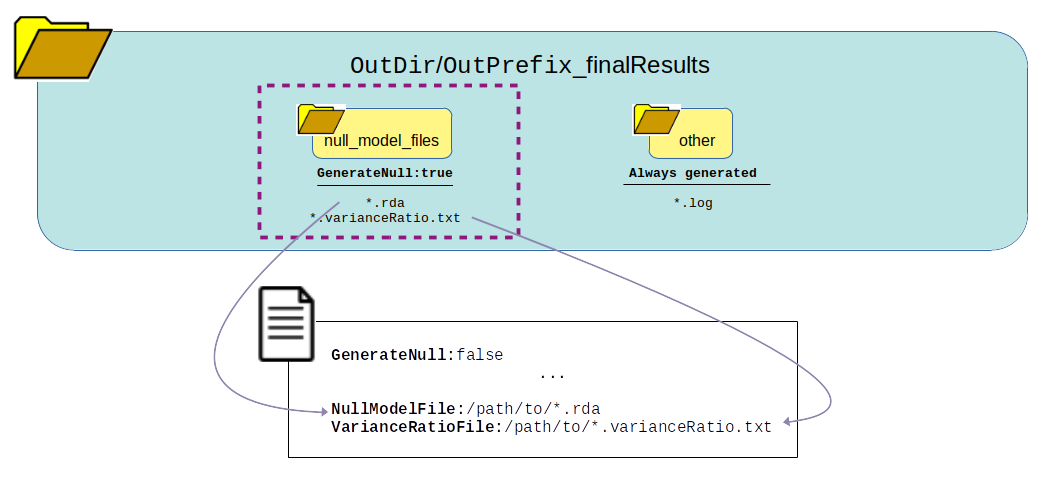
What can be reused?
Association Analysis only¶
Setting this logic (see OPTION A: Imputation files need window chunking):
GenerateGRM:false
GenerateNull:false
GenerateAssociations:true
GenerateResults:false
SkipChunking:false
SaveChunks:true
Returns this output:
What can be reused?
Setting this logic (see OPTION B: Imputation files do not need window chunking and will be reused):
GenerateGRM:false GenerateNull:false GenerateAssociations:true GenerateResults:false SkipChunking:true
Returns this output:
What can be reused?
Output Files Generated¶
This section describes the file format output and what is located in each result file.
For association analysis results file per snp. This is for text files generated with the suffix: * *_allSNPs_noFiltering.txt.gz * *_commonSNPs_cleaned.txt.gz * *_rareSNPs_cleaned.txt.gz
Column |
Interpretation |
|---|---|
CHR |
chromosome the variant is located |
POS |
base position of the variant (hg19 vs hg38 is based upon what was specified at runtime) |
majorAllele |
major allele (most common frequency in the input population) |
minorAllele |
minor allele (lesser common frequency in the input population) |
SNPID |
variant name |
BETA |
effect size based on the minor allele |
SE |
standard error of beta |
OR |
odds Ratio |
LogOR |
log(odds ratio) |
Lower95OR |
lower 95% confidence interval of the odds ratio |
Upper95OR |
upper 95% confidence interval of the odds ratio |
MAF |
minor allele frequency |
MAC |
minor allele count |
p.value |
p-value of the variant |
N.Cases |
number of cases evaluated for this variant (binary traits only) |
N.Controls |
number of controls evaluated for this variant (binary traits only) |
casesHomMinor |
number of cases evaluated for this variant that are homozygous for the minor allele (binary traits only) |
casesHet |
number of cases evaluated for this variant that are heterozygous (binary traits only) |
controlHomMinor |
number of control evaluated for this variant that are homozygous for the minor allele (binary traits only) |
controlHet |
number of controls evaluated for this variant that are heterozygous (binary traits only) |
negLog10pvalue |
negative log base 10 of the p-value |
R2 |
imputation quality for imputed variants |
ER2 |
empirical R2 quality extracted from info file from imputation – for genoytyped variants only |
GENOTYPE_STATUS |
imputed, genotyped or genotyped and imputed status for the variant |
There is an intermediate file also output with the suffix allChromosomeResultsMerged.txt. This is output directly from SAIGE,
however, it is not cleaned and alleles and betas have not been corrected for minor allele basis. All GWAS results, including the graphs
generated from the pipeline are based on the files above ending in _commonSNPs_cleaned.txt.gz and _rareSNPs_cleaned.txt.gz.
However, some T statistics and variance calculations are included in this file, along if the variant converged or not. Therefore, it is still saved in the output so it can easily be searched and referenced for particular variants, if needed.
Column |
Interpretation |
|---|---|
CHR |
chromosome the variant is located |
POS |
base position of the variant |
SNPID |
variant name |
Allele1 |
allele 1 (careful, not necessarily major allele) |
Allele2 |
allele 2 (careful, not necessarily minor allele) |
AC_Allele2 |
allele count of allele 2 |
AF_Allele2 |
allele frequency of allele 2 (not necessarily minor allele) |
imputationInfo |
imputation quality score |
N |
total samples considered in model for this variant |
BETA |
effect size of allele 2 (not necessarily minor allele) |
SE |
standard error of beta |
p.value |
p-value of the variant |
p.value.NA |
p-value of variant when SPA is not applied |
Is.SPA.converge |
whether saddle point approximation (SPA) converged for this variant |
Tstat |
estimated variance of score statistic with sample related incorporated |
varTstar |
variance of score statistic without sample related incorporated |
AF.Cases |
allele frequency of allele 2 in cases (only for binary trait) |
AF.Controls |
allele frequency of allele 2 in controls (only for binary traits) |
N.Cases |
total cases considered in model for this variant (only for binary traits) |
N.Controls |
total controls considered in model for this variant (only for binary traits) |
homN_Allele2_cases |
homozygote counts in cases |
hetN_Allele2_cases |
heterozygote counts in cases |
homN_Allele2_ctrls |
homozygote counts in controls |
hetN_Allele2_ctrls |
heterozygote counts in controls |